|
|
The Sickbed, Medieval Illum MS. |
|
|
The Sickbed, Medieval Illum MS. |
Images and pathol. adapted from: https://www.nejm.org/doi/full/10.1056/NEJMicm1710121
A CATHOLIC priest asked for assistance concerning a patient he had been asked to visit in a hospice/palliative care facility near his parish. The patient was a 32 year old woman diagnosed with Kreutzfeldt-Jakob disease, an untreatable, relentlessly progressive form of presenile dementia. As her condition deteriorated she had been moved from home-care to an extended care facility, and was now receiving hospice/palliative care. She was unable to take food by mouth and unable to communicate, although she would sometimes grasp fingers placed in her hand. She was receiving no medication or treatment other than an intravenous line supplying her with fluids and morphine.
The priest was concerned that, although he had been told she was expected to die soon, she appeared unchanged throughout a two-week period during which he had visited her. A visitor who described herself as a “pro-life nurse” (not a member of the staff of the extended care facility) advised the priest that a second neurological consultation should be obtained (“from a pro-life neurologist”) and that the use of morphine should be reconsidered, “since it might be intended to hasten her death”.
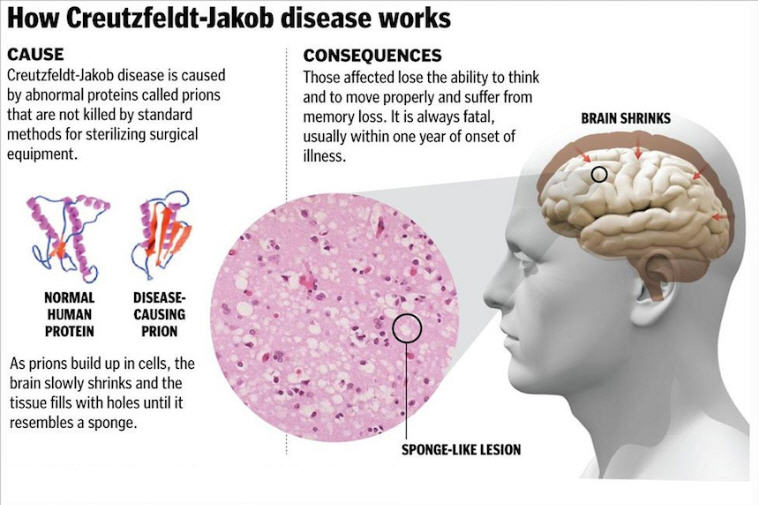
CREUTZFELDT –Jakob disease [is], a fatal spongiform encephalopathy that is caused by the accumulation of abnormal prions (helical proteins of unknown function). Creutzfeldt–Jakob disease leads to cognitive decline and is characterized by additional neurologic features that manifest according to the location of lesions. The disease is always fatal, usually within one year of diagnosis.
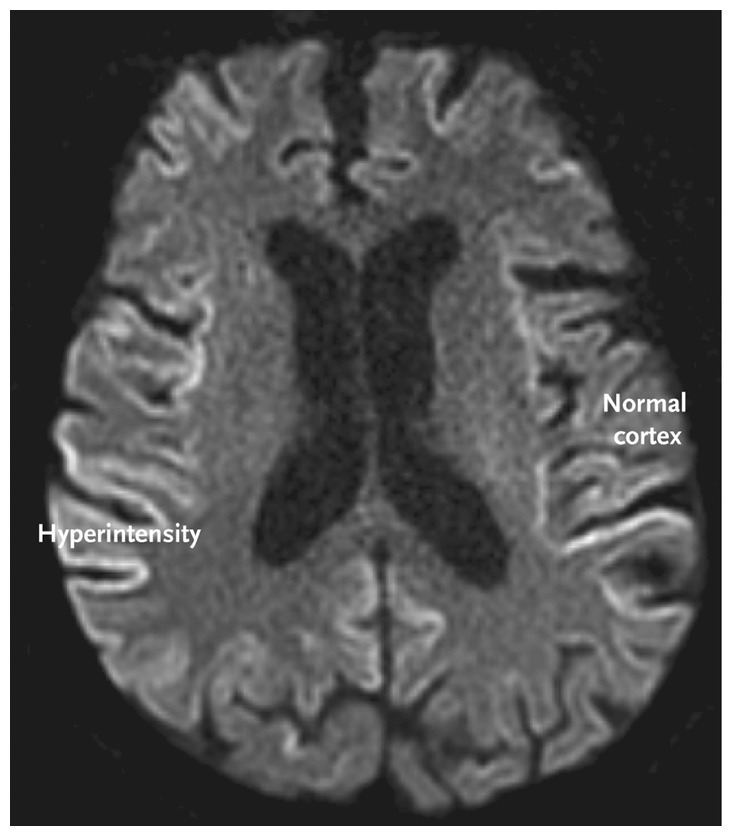
Common MRI findings include hyperintensity of the cortex, pulvinar and dorsomedial thalamic nuclei, and basal ganglia on T2-weighted, diffusion-weighted, and fluid-attenuated inversion recovery sequences.
|
|
[...] CREUTZFELDT –Jakob disease is the most common of the human prion diseases, a group of neurodegenerative disorders with unique pathogenesis and means of transmission. Prion diseases
may arise sporadically,
be transmitted as infectious diseases,
or be inherited as genetic disorders.
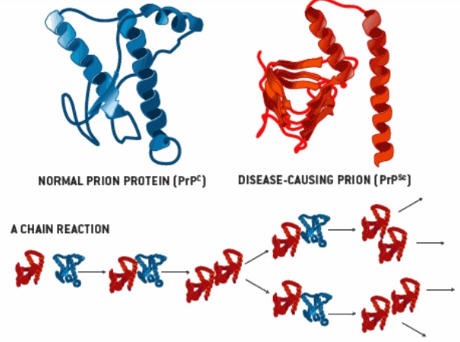
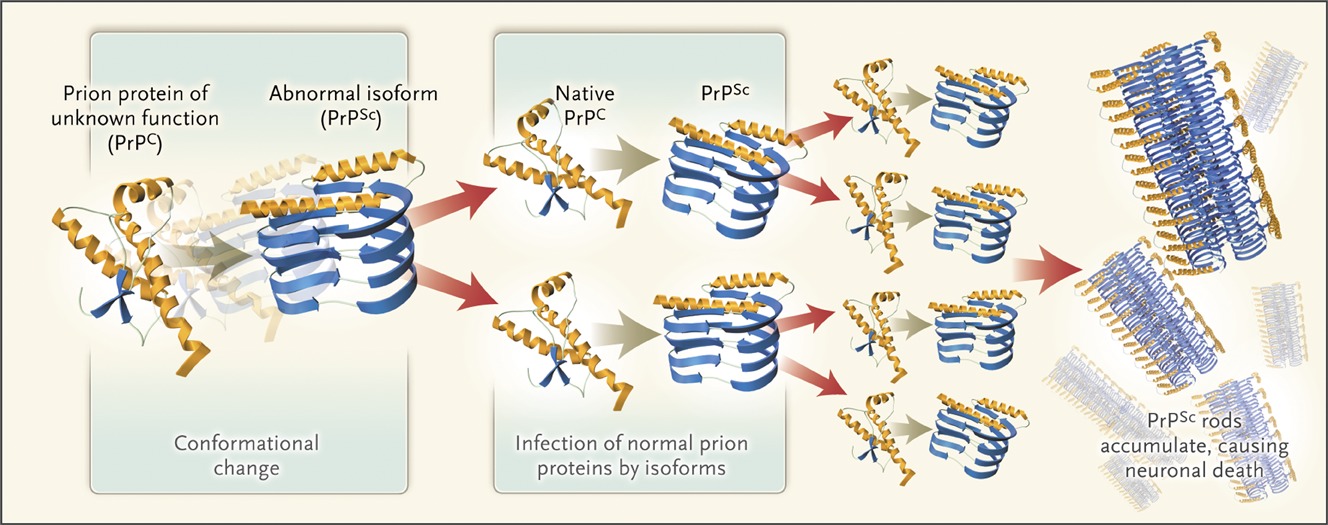
The underlying cause is the conversion of a normal cellular prion protein of unknown function (PrPC) to an abnormal isoform (PrPSc) by means of a conformational change. This abnormal protein is capable of inducing the same structural change in native prion proteins, which accounts for its infective capacity. Conformationally altered proteins accumulate within neurons, eventually causing neuronal death . Widespread neuronal loss leads to vacuolization (spongiform changes) in the cortex and the clinical manifestations of subacute spongiform encephalopathy.
|
|
|
|
[...] CREUTZFELDT –Jakob disease is the most common of the human prion diseases, a group of neurodegenerative disorders with unique pathogenesis and means of transmission. Prion diseases
may arise sporadically,
be transmitted as infectious diseases,
or be inherited as genetic disorders.
The underlying cause is the conversion of a normal cellular prion protein of unknown function (PrPC) to an abnormal isoform (PrPSc) by means of a conformational (structural-shape) change. This abnormal protein is capable of inducing the same structural change in native prion proteins, which accounts for its infective capacity. Conformationally altered (abnormally folded) proteins accumulate (clump in aggregates) within neurons, eventually causing death of neurons (brain cells). Widespread neuronal loss leads to vacuolization (spongiform changes) in the cortex (surface of the brain) and the clinical manifestations of subacute spongiform encephalopathy.
|
|
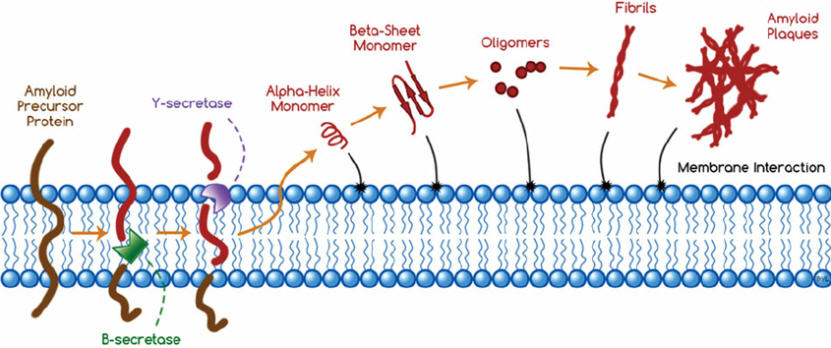
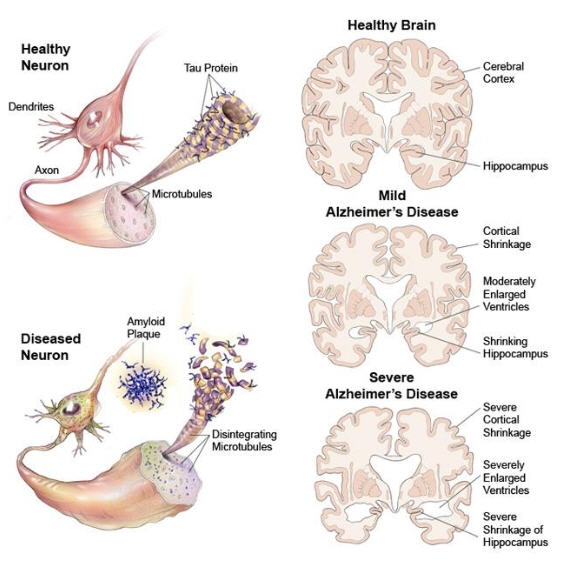
ABNORMAL protein aggregates (β-amyloid plaques) may accumulate through a different mechanism in more common forms of dementia, such as Alzheimer's disease.
|
|
|
|
This Webpage was created for a workshop held at Saint Andrew's Abbey, Valyermo, California in 1998